
Фолдинг есть, но на уровне третичной структуры белка. На уровне вторичной структуры его нет. Программная аллотропия белков опровергает гипотезу фолдинга
Технология трансляции генетического кода в структуру белка
Высоко периодичные структуры белков
Письмо, которое нужно разослать по лабораториям РСА
В лаборатории Наномир создана новая технология определения структуры белковых молекул, которая работает примерно в миллиард раз быстрее РСА. Наша задача в кратчайшие сроки проверить новую технологию, т.к. от её внедрения человечество может получить фантастический эффект.
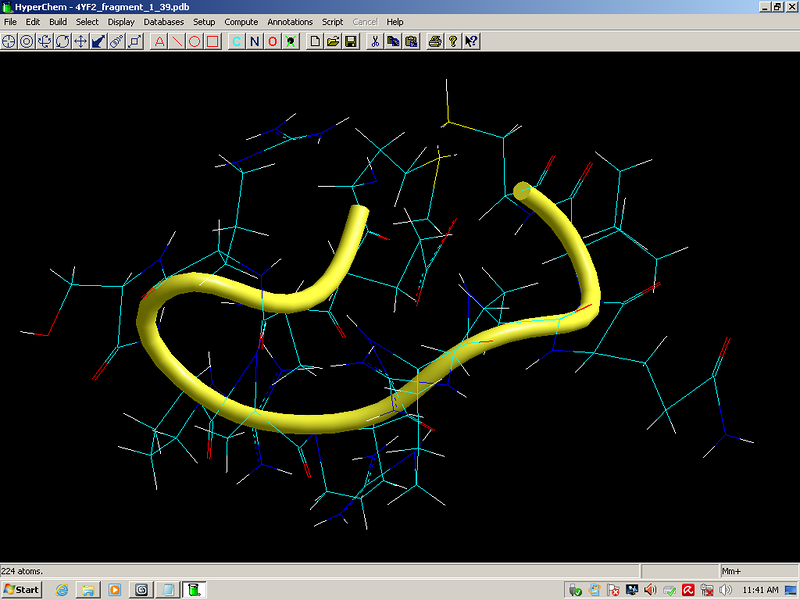
Проще всего проверить новую технологию на простых структурах типа поливалина:
https://img-fotki.yandex.ru/get/509292/ … 5_orig.png
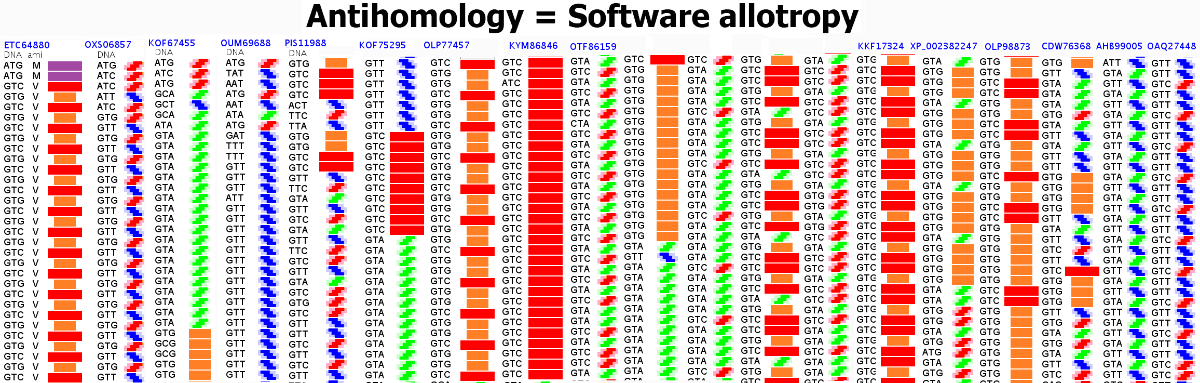
ETC64880: https://www.ncbi.nlm.nih.gov/nuccore/564568708
OXS06857: https://www.ncbi.nlm.nih.gov/nuccore/1226034760
KOF67455: https://www.ncbi.nlm.nih.gov/nuccore/918287724
OUM69688: https://www.ncbi.nlm.nih.gov/nuccore/1198313040
PIS11988: https://www.ncbi.nlm.nih.gov/nuccore/1277217132
KOF75295: https://www.ncbi.nlm.nih.gov/nuccore/918302080
OLP77457: https://www.ncbi.nlm.nih.gov/nuccore/1129165616
KYM86846: https://www.ncbi.nlm.nih.gov/nuccore/1009361734?&fmt_mask=65536
OTF86159: https://www.ncbi.nlm.nih.gov/nuccore/CM007906.1?report=fasta&sat=37&satkey=307832265&itemID=1362
KKF17324: https://www.ncbi.nlm.nih.gov/nuccore/808866584?&fmt_mask=65536
XP_002382247: https://www.ncbi.nlm.nih.gov/nuccore/238502026
OLP98873: https://www.ncbi.nlm.nih.gov/nuccore/1129194414
CDW76368: https://www.ncbi.nlm.nih.gov/nuccore/678335661
AHB99005: https://www.ncbi.nlm.nih.gov/nuccore/564747871?&fmt_mask=65536
OAQ27448: https://www.ncbi.nlm.nih.gov/nuccore/1032646064
OUM69730: https://www.ncbi.nlm.nih.gov/nuccore/1198313040
OGL53990: https://www.ncbi.nlm.nih.gov/nuccore/1084158629
EIE92391: https://www.ncbi.nlm.nih.gov/nuccore/76151980
XP_001590524: https://www.ncbi.nlm.nih.gov/nuccore/156049114
OTG33229: https://www.ncbi.nlm.nih.gov/nuccore/CM007891.1?from=7600734&to=7601591&sat=37&sat_key=307832447
OLQ06943: https://www.ncbi.nlm.nih.gov/nuccore/1129204435
XP_001895277: https://www.ncbi.nlm.nih.gov/nuccore/170580473
XP_001584576: https://www.ncbi.nlm.nih.gov/nuccore/156030497
KOF84884: https://www.ncbi.nlm.nih.gov/nuccore/918315370
Возможно ли методом РСА определить структуру этих участков белковых молекул? Если да, то нужен прайс.
С уважением, Руководитель лаборатории Наномир, Александр Кушелев
2017.12.14 13:23:02
Кушелев Александр Юрьевич: Мы с Викторией соколик стали обсуждать фолдинг, и тут выяснилось, что она перепутала углы поворота транспортной РНК и белковой спирали
Отсюда и странные углы спиралей...
2017.12.08 11:31:07
активное втюхивание того, что может сделать программа Кушелева (2D-схему белка), вместо того, что хотят видеть заказчики 3D-модель белка, не приведёт к желаемому результату.
Кушелев: Тут есть ряд нюансов. Во-первых, нужно объяснить всем, кто хочет видеть 3D-модели белков, что рентгеноструктурный анализ (РСА) в действительности не может достоверно показать даже вторичную структуру белка. Что касается третичной и четвертичной, то РСА показывает их условно, т.е. взаимное расположение неких крупных ассоциаций, например, спиральных участков. При этом РСА часто не может отличить разные типы спиралей, например, пи-спираль от альфа-спирали, альфа-спираль от 310-спирали. А большинство спиральных участков гибридные, т.е. представлены чередованием композиций альфа- и 310-спиралей. С помощью РСА об этом не удалось узнать за многие десятилетия исследований. Поэтому доверие к интерпретации данных РСА держится на недостатке знаний в этой области, т.е. грубо говоря на невежестве профессионалов по молекулярной биологии
Во-вторых, программа Пикотех 3D позволяет показать и проконтролировать достоверность 3D моделей для некоторых участков белковых молекул. Например, для циклов, замкнутых через дисульфидные мостики. Речь идёт о циклах лизоцимов, где методом структурного анализа надежно установлены факты замыкания циклов через дисульфидные мостики:

http://nanoworld88.narod.ru/data/285.htm
http://nanoworld88.narod.ru/data/586.htm
Цитата: В общей сложности построены модели 6 замкнутых через дисульфидные мостики участков.
1. Cys 95 - Cys 113 в человеческом лизоциме.
2. Cys 96 - Cys 117 в альтернативном человеческом лизоциме.
3. Cys 78 - Cys 82 в альтернативном человеческом лизоциме.
4. Cys 94 - Cys 98 в лизоциме куриного яйца
5. Met 1 - Cys 13 в мышином лизоциме
6. Met 1 - Cys 24 в лизоциме куриного яйца
Другой пример - длинные фундаментальные (альфа-, 310-, пи-) и программные спирали:

Кушелев: Если Вы признаёте существование программных спиралей, то
1. Во-первых, почему они не изображаются у Вас на уровне 2D-структуры?
2. О каком фолдинге может идти речь, если 3D модель программных спиралей строится по таблице композиционного кода?
Victoriy пишет:
Специфика 3/10, пи- и альфа-спиралей формируется уже в ходе процесса фолдинга на основе простейшего структурного шаблона 3/10 спирали.
Кушелев: А тут подробнее, пожалуйста.
Вот вторичная структура двух полилизинов:
Вот, что показывает для пи-спирали алгоритм Кушелева и Соколик:

Ваш алгоритм 2D ничего не показывает. Откуда при 3D моделировании у Вас появляется пи-спираль? Каков конкретный алгоритм Вашей 3D программы, который строит пи-спираль по коду:
>XM_022674627.1 PREDICTED: Astyanax mexicanus protein FAM133-like (LOC111193520), partial mRNA
GTCCAGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAA
Victoriy пишет:
декодированные спиральные структуры белка не обязательно сохраняются в полном объёме в его функциональной конформации после фолдинга: нередко они полностью трансформируются в бета-тяжи и петлевые структуры/повороты. Пересмотрите примеры в монографии "Пикотехнология белков", там много таких белков.
Кушелев: Вот мне и интересно, как эти два белка (пи-спиральный и альфа-спиральный) моделируются в Вашей программе. Можно пошагово показать, как по коду n(AAA) Ваша программа получает пи-спираль, а по коду n(AAG) - альфа-спираль. И ещё очень интересно посмотреть Вашу 3D модель программной 35-спирали, когда коды AAA и AAG чередуются. По моей таблице композиционного кода получается такая модель:

Пример программной 35-спирали (компактное 2D-представление)
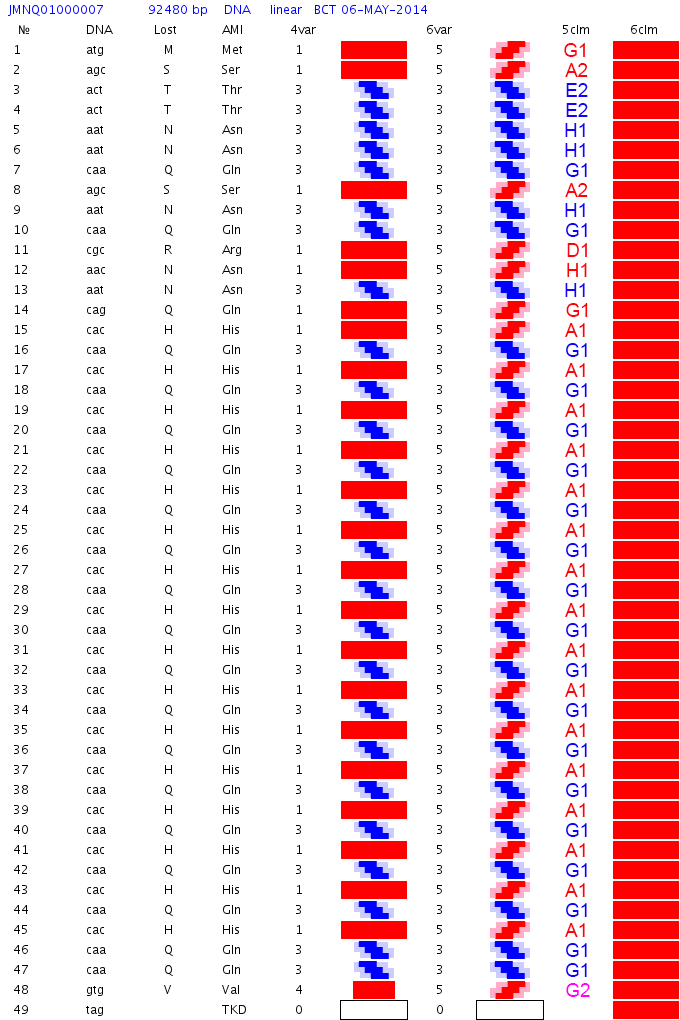
Пример программной 35-спирали (развернутое 2D-представление)

Подробнее: https://subscribe.ru/archive/science.ne … 3210.html/
3D-модель белковой молекулы с программной 35-спиралью.
2017.12.08 11:48:10
Victoriy пишет:
программа "Молекулярный конструктор" Виктории Соколик способна выдавать как 2D-схему, так и 3D-модель структурного шаблона белка по детерминирующей его нуклеотидной последовательности.
Кушелев: Это очень здjрово. Но хотелось бы уточнить, как конкретно, т.е. по шагам, получается, например, 3D модель этого белка:
2D структуру алгоритм, реализованный в программе Prosolver не показывает, но и "не очень-то хотелось". Интересно посмотреть по шагам, как программа Молекулярный конструктор строит 3D-модель этого белка, т.е. пи-спираль. Ну и сравнить с постройкой альфа-спирали из тех же остатков лизина.
Сможете показать по шагам работу алгоритма?
2017.12.08 12:01:24
Victoriy пишет:
Единственно что необходимо для полноценного удовлетворения заказчика, это виртуально в соответствующих программах (типа молекулярная динамика - МД) оптимизировать полученную структуру шаблона белка с учетом ближнего физ-хим. окружения его молекулы, т.е. смоделировать его фолдинг как бы в природных условиях, но виртуально.
Кушелев: Гипотеза фолдинга опровергается программной аллотропией (композиционным кодированием) структуры белка:

Что касается программ оптимизации, то стандартные программы не учитывают кольцевую форму электрона, поэтому непригодны для оптимизации пикотехнологических моделей.
2017.12.08 12:13:15
Victoriy пишет:
... любая заказываемая модель белка необходима не просто так для красоты и любопытства, а для её дальнейшего изучения на предмет эффективности связывания (образования комплекса) с целевыми лигандами - виртуального докинга. Последний шаг и есть практическое внедрение в фармакологии, биотехнологии и др. областях.
Поэтому на этапах молекулярная динамика/докинг лаборатории Наномир понадобятся услуги опытного молекулярного биолога либо специального он-лайн сайта (такие принимали участие в CASP), а не кучи программистов, о которых говорит А.Ю. Либо же предлагать лабораториям структурный шаблон белка и оговаривать, что они должны над ним ещё сами поработать вышеописанным способом (обычно специалистов по МД и докингу в таких лабораториях достаточно).
Кушелев: Конечно, можно пытаться оптимизировать пикотехнологическую модель нанотехнологическими алгоритмами, но это неэффективно. Это примерно то же самое, что собирать айфон методом лепки из глины.
Для тех, кто не видит картинки, я буду подписывать: "Айфон, слепленный из пластилина".
Другое дело - создать программы оптимизации пикотехнологического уровня. Это безусловно сложнее, но и результат будет соответствующий:
Цикл лизоцима, замкнутый через дисульфидный мостик.
Нужно обратить внимание на то, что 3D модели некоторых участков белков получаются с пикотехнологической точностью даже без оптимизации, т.е. на уровне геометрического алгоритма. Согласитесь, что точность на уровне пикометров впечатляет по сравнению с тем, что РСА не может отличить пи-спираль от альфа-спирали.
2017.12.11 11:50:23
aest пишет:
А к чему вся эта дискуссия по альфа спиралям сейчас ?
Кушелев: А при том, что альфа-спираль является жесткой структурой. Ее можно повернуть на суставе Pro относительно другой спирали:
А Виктория предлагает менять вторичную структуру. А это равносильно "размягчению костей"

2017.12.11 12:24:23
Victoriy пишет:
Обратите Ваше пристальное внимание на то, что углы композиции между аминокислотными остатками задаются внутри рибосомы, а вот водородные связи, дополнительно фиксирующие их взаимное расположение уже на выходе из неё.
Кушелев: С чего бы это? Если аминокислота соединена с предыдущей, водородная связь образуется за миллисекунду:

Подробнее о протонной релаксации водородных связей: http://crm-en.ics.org.ru/uploads/kim3/crm09307.pdf
Victoriy пишет: В рибосоме водородные связи не образуются, только на выходе из неё за те же миллисекунды.
Кушелев: С чего Вы это взяли? В канале рибосомы умещаются даже все радикалы альфа-спирали. Это значит, что там та же среда, что и за пределами рибосомы. Те же молекулы воды, протоны, образующие водородные связи. Как только присоединена очередная аминокислота (0.2 ... 0.6 сек), так уже через миллисекунду образуется водородная связь, т.е. до установки следующей аминокислоты. Другого варианта нет.
Victoriy пишет: Вы уже заложили свою таблицу композиционного кода и углы для альфа-спирали и Вам проще отстаивать эту ошибку
Кушелев: Зачем отстаивать? Эксперимент показывает, что для замыкания дисульфидных мостиков фолдинг не нужен
Цикл, собранный по геометрическому алгоритму замыкается и без него:
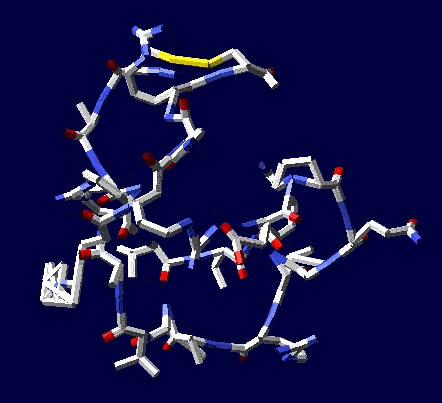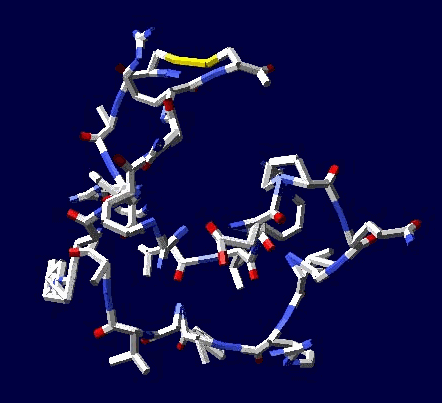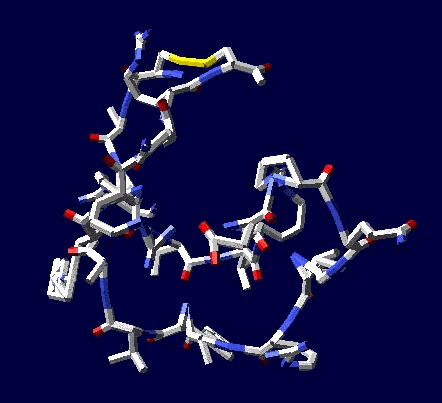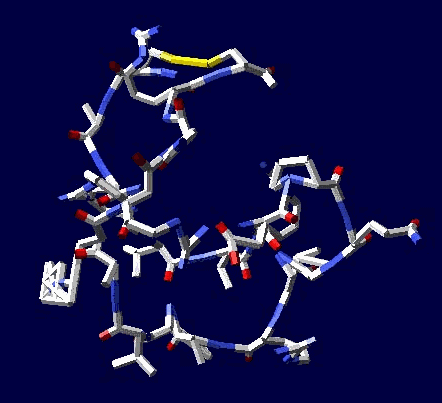
2017.12.09 20:14:49
Кушелев: Изменение нескольких углов может привести к тому, что цикл из 22 аминокислот не замкнется вообще, т.е. не с вероятностью 1/30 000 000 000, а с вероятностью строго равной нулю (=0).
Поэтому я и пишу, что замыкание 6 циклов из разных лизоцимов не может быть случайностью. Эти замыкания показывают, что таблица композиционного генетического кода и композиционные углы правильные. Не все, конечно, а только те, которые принимают участие в формировании этих циклов.
2017.12.11 12:37:24
Victoriy пишет:
статистический анализ приведенный в монографии "Пикотехнология белков", на основе которого сделана моя таблица композиционного кода, не показал специфику кодирования альфа- и 3/10 спиралей разными кодонами. Вашей статистики на этот счет нет, поэтому такое утверждение голословно, и ненаучно его отстаивать.
Кушелев: Если анализ не показал, то это не означает, что повторный анализ не покажет
Мы с Вами знаем, что специалисты по рентгеноструктурному анализу редко отличают 310-спираль от альфа-спирали. Более того, за многие десятилетия существования РСА протяженные участки пи-спиралей вообще оказались незамеченными...
А они есть!

Так что в следующей книге "Пикотехнология белков-2" у Вас есть возможность продемонстрировать и 6 фрагментов лизоцимов, замкнутых через дисульфидные мостики без фолдинга, и сверхдлинные фундаментальные и программные спирали белковых молекул, структура которых определена по таблице композиционного кода, где Ваши данные для альфа- и пи-спиралей полностью совпадают с моими. Вы ещё хотели показать экспериментальные данные, с которыми совпадают Ваши альфа- и пи-спирали длиной более 63 витков:

Оригинал: https://img-fotki.yandex.ru/get/892397/ … 5_orig.png
А если этих экспериментальных данных еще нет, то что мешает их получить? В институте белка мне сообщили, что определить структуру с помощью РСА могут в Курчатовском институте. Давайте вместе предложим им определить две структуры полилизина, закодированные триплетами n(AAG) и n(AAA).
2017.12.09 20:50:04
Кушелев: На иллюстрации показано, что вторичная структура, построенная по алгоритму Виктории Соколик на 100% совпадает со вторичной структурой, полученной по композиционной таблице Кушелева. Это только в программе от Prosolver коду AAA ничего не соответствует, а по алгоритму Виктории Соколик код AAA соответствует левой спирали, т.е. пи-спирали. Вам осталось подправить углы пи-спирали, чтобы не пришлось получать "те же результаты другим способом" уже в 3D версии программы
Если я не ошибаюсь, Вы предлагаете строить модель пи-спирали в два этапа. На первом построить неправильную левую спираль (3 АО на виток), т.е. с неправильными водородными связями, соединяющими n-ый АО с (n+3)-им, а на втором этапе переделать неправильную левую спираль в правильную пи-спираль, т.е. разорвать все водородные связи n ... (n+3), изменить все углы между аминокислотными остатками со 120 градусов до 87, после чего сделать заново все водородные связи, но уже не с третьим АО, а с пятым, т.е. n ... (n+5). Другими словами, неправильно построить, потом всё сломать и построить правильно. А почему нельзя сразу сделать правильно по Вашей же таблице композиционного кода, которая в этом месте на 100% совпадает с моей? Просто сразу взять правильные углы пи-спирали.




Альфа-спираль Бета-спираль Пи-спираль 310-спираль
Подробнее можно посмотреть в 287-ом выпуске рассылки: http://nanoworld88.narod.ru/data/287.htm
2017.12.10 14:04:17
Когда Виктория Соколик, писала книгу "Пикотехнология белков", научных открытий, о которых сегодня идет речь, ещё не было. Поэтому эти иллюстрации не могли попасть в книгу из будущего:
Но буквально вчера выяснилось, что в программе от Prosolver вторичная структура показана не так, как должна быть показана согласно таблице композиционного кода от Виктории Соколик.
Там, где программа показывает отсутствие вторичной структуры, в действительности по таблице Виктории Соколик вторичная структура есть. Это структура левой спирали, т.е. пи-спирали:
Так что разногласий во вторичной структуре нет. Разногласие в величине угла, т.е. на уровне пространственной модели, т.е. 3D версии программы.
Виктория считает, что рисобома собирает левую спираль с углами по 120 градусов (три АО на виток), после чего некий "фолдинг" ломает эту структуру, т.е. разрывает водородные связи n ... (n+3), меняет все углы со 120 на 87 градусов и создаёт все водородные связи заново, т.е. уже n ... (n+5). А я считаю, что рибосома строит пи-спираль с первого раза. Без мистического фолдинга.
2017.12.11 12:45:51
aest пишет:
лизоцим не является жесткой фигурой, подобной треугольнику, там вполне возможно согласованно поизменять углы без разрыва дисульфидного мостика, подобно тому, как это можно сделать в параллелограмме
Кушелев: Только он замкнулся через дисульфидный мостик без изменения углов. А это значит, что фолдинга нет на уровне вторичной структуры. Кстати, Вы можете "поизменять углы" и выложить на форум результат своего колдовства. Интересно посмотреть, что за углы Вы будете менять, и удастся ли Вам поменять их согласованно?
Ваши "просто слова" против модельного эксперимента ровным счётом ничего не значат
Сначала измените согласованно углы, но не в параллелограмме, а в модели фрагмента лизоцима, а мы посмотрим, замкнётся ли он после этого или нет.
2017.12.11 13:08:24
Вы строите свою таблицу кодирования разновидностей спиралей опираясь на свои фантазии и отмахиваясь от результатов статистики.
Кушелев: Вообще-то я сразу показал, как я строил свою таблицу композиционного генетического кода
Я воспользовался данными РСА для наиболее изученных белков. При этом на пластмассовых моделях я получил параметры альфа-спирали, 310-спирали, пи-спирали и бета-спирали. Модельные эксперименты показали, что составленная таблица композиционного кода работает без ошибок. Позднее, когда сборка моделей белка была автоматизирована, обнаружился цикл лизоцима, замкнувшийся через дисульфидный мостик без фолдинга, т.е. строго по таблице композиционного кода.
Ещё позднее я нашел ещё 5 фрагментов разных лизоцимов, замнувшихся без фолдинга.
Более того, в процессе этого исследования я обнаружил замыкание дисульфидных мостиков не только между цистеинами, но и между цистеином и метионином, что явилось научным открытием, согласитесь. Вам известно, что дисульфидные мостики могут образовываться между цистеином и метионином?
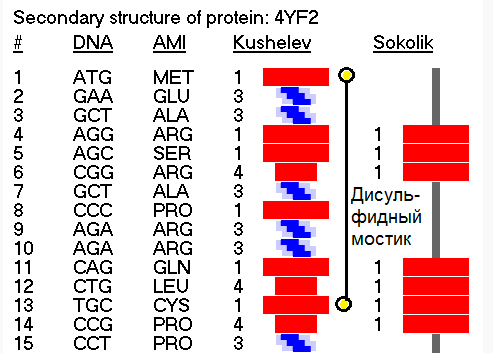
Здесь программа от Prosolver показывает отсутствие вторичной структуры по композиционному коду пи-спирали, но мы уже выяснили, что таблицы композиционного кода по части пи-спирали совпадают, поэтому в действительности эти циклы лизоцима обязаны замкнуться и по алгоритму Виктории Соколик
Кстати, эти модели совпадают и с экспериментальными данными, если не считать дисульфидных мостиков между цистеином и метионином. Но тут уж дело времени. Должны появлиться экспериментальные данные и по дисульфидным мостикам между метионином и цистеином. Рано или поздно экспериментаторы должны обнаружить эти мостики...
2017.12.11 13:34:23
Меняйте согласованно углы. Но не забывайте, что эти углы не лежат в одной плоскости
aest пишет: Возьмите металлическую цепь о тысячи звеньях, замкните. Бросьте комком на землю. Сколько там углов между звеньями согласованно изменились не лежа при этом в одной плоскости? Цепь не разомкнулась. Мистика...
Кушелев: Жаль, что Вы не отличаете механизм белковой молекулы от механизма цепи. В молекуле белка вращение происходит вокруг некоторых осей:

В учебнике молекулярной биологии есть такой термин: "вращение затоможено". Это определяется как раз структурой электронной оболочки молекулы. Так же, как рука может гнуться в локте не по любому направлению. Сустав - это не шарнир.
2017.12.11 14:54:25
абсолютно не соответствующие реальному механизму трансляции белка.
А соответствует очередь протонов на образование водородных связей в альфа-спирали на выходе из канала рибосомы?
Кушелев: Вы согласны, что в канале рибосомы присутствуют протоны? Если согласны, то что им мешает через миллисекунду после установки очередного аминокислотного остатка образовать водородную связь? Т.е. ещё до того, как тРНК отпустит аминокислотный остаток, который она держит 4-мя вандерваальсовыми связями:

Подробнее: http://nanoworld88.narod.ru/data/216.htm
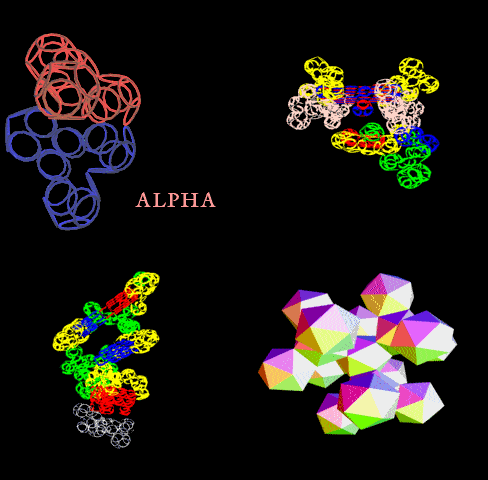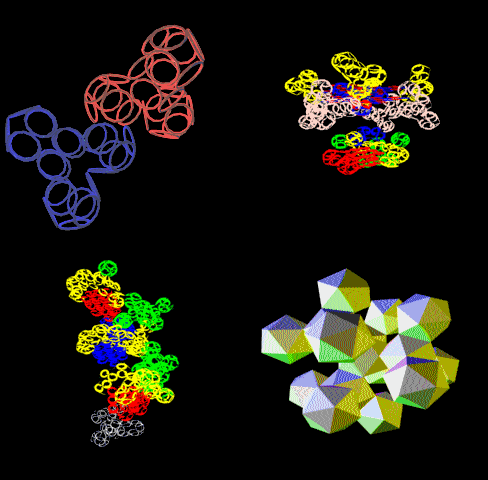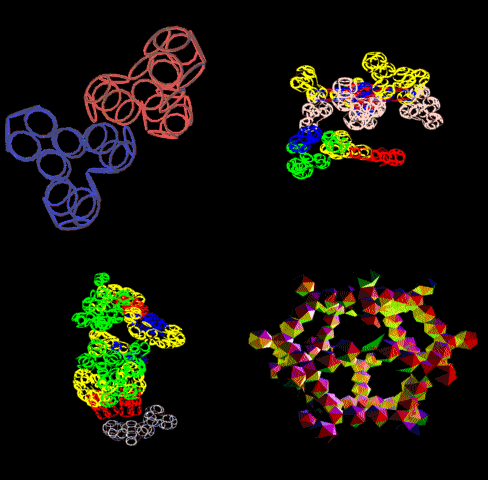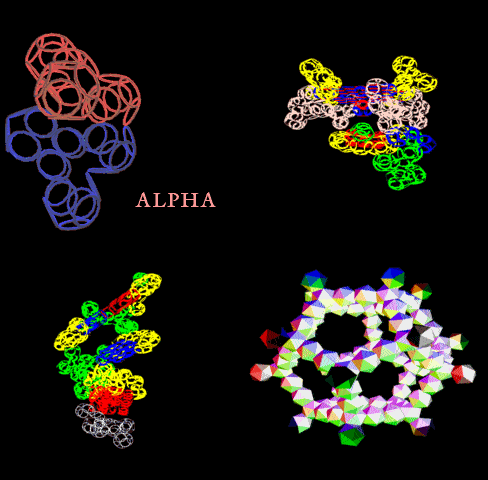
Подробнее: http://nanoworld88.narod.ru/data/247.htm
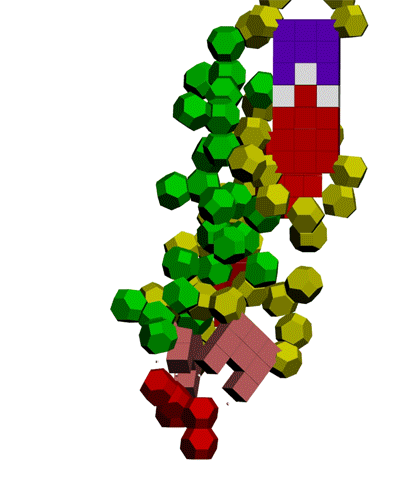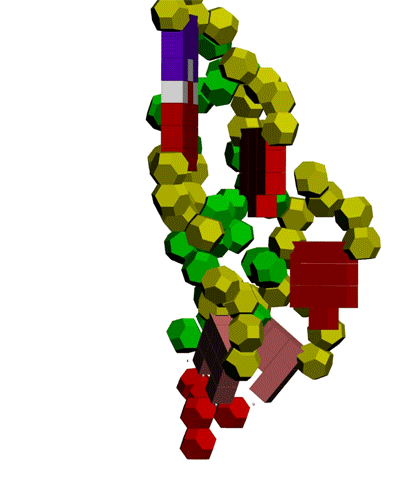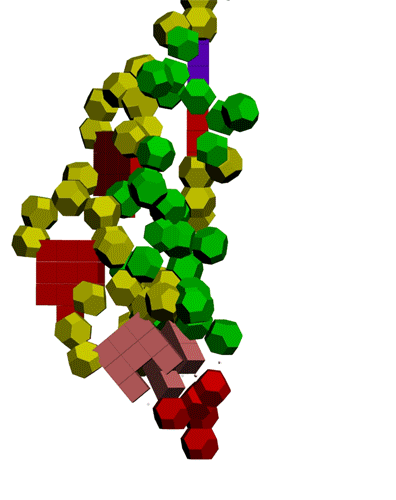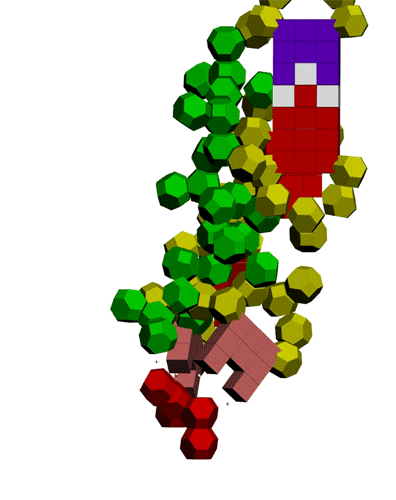
2017.12.11 19:36:34

Кушелев: Судя по этой схеме, Виктория отмеряет углы поворота tRNA вокруг оси белковых спиралей.
Увжаемая Виктория! А Вас не смутил угол 0 градусов? Или Вы его "смело заменили" на 180 градусов?
Кстати, эту ошибку легко исправить.
Достаточно организовать "фолдинг" таблицы композиционных углов от Виктории Соколик, чтобы получилась таблица композиционных углов от Александра Кушелева.
Сами структуры белков фолдировать не нужно. Достаточно фолдировать таблицу углов
А я никак не мог понять, откуда Вы такие углы берете? Это можно назвать "антифолдинг" таблицы композиционных углов. Осталось добавить "фолдинг" таблицы композиционных углов, "и всех делов" .
Получается, что Вы не показывали Prosolver иллюстрации, где показано, вокруг каких осей вращаются модели аминокислотных остатков?
Есть же описание алгоритма от Евгения Неделько, которое находится в открытом доступе с 1998 года.
В программе Неделько выводились только координаты центров атомов, а углы были взяты у Рамачандрана. Это уже в более поздней версии Дениса Савина программа скрипт стала выводить не только координаты центров атомов, но и кольцегранные оболочки. Помню Вы там ещё ошибку нашли. Правда, программа правильно заработала после исправления второй ошибки. Оказалось, что Денис Савин перепутал в одном месте координату в уравнениях. Потом мы с Валентином поменяли зеркальные отражения аминокислотных остатков на правильные и промасштабировали модели по экспериментальным данным.
Кстати, в программе Дениса Савина вращение тоже реализовано не вокруг одной оси, как было бы естественно, а последовательно вокруг трёх декартовых осей. Это уже связано с вычислительной средой. При этом сначала оказалось, что все спирали строятся правильно, а изломы спиралей неправильно. Выяснилось, что правильные углы между спиралями разных типов получаются при правильном задании начала отсчета углов. Другими словами, нужно поставить аминокислоту в правильную позицию, правильно сориентировать по всем трем осям, после чего получаются корректные модели белков. Но это только геометрический уровень.
2017.12.11 09:59:42
aest пишет:
Вы приводите в качестве примеров жесткие фигуры, у которых грани как раз треугольники. Но масса таких фигур ничтожна по сравнению с массой нежестких фигур. Так что основной, имеющий важность для науки вывод не опровергнут: фолдинг есть. А вы можете и дальше его не замечать, пусть он тут для других зияет
Кушелев: Вы очень настойчиво не хотите изучать азы философии. Кстати, Ваше заявление по поводу "массы таких фигур", применительно к белкам, тоже ошибочно. Подавляющее большинство белков содержат альфа-спиральные участки. На этих иллюстрациях они показаны красным цветом. Их можетбыть много, может быть мало.


Альфа-спиральные участки присутствуют в подавляющем большинстве белков. Их отсутствие является редким исключением. Так что Вам срочно нужна новая ошибка "для оправдания"

Попобуйте, поколдуйте, например, над жёсткостью альфа-спирали.
2017.12.11 10:15:37
aest пишет:
фолдинг происходит благодаря наличию у белка "суставов", т.е. мест, где он легко может гнуться, о чем и говорит Виктория. Так что, повторюсь, основной, имеющий важность для науки вывод не опровергнут: фолдинг есть, а пикотехнология Кушелева ошибочна
Кушелев: Вы, вероятно, не поняли моего объяснения. На суставах может гнуться третичная структура белка, т.е. состоящая, например, из жёстких альфа-спиральных участков. Действительно, на уровне третичной структуры есть и фолдинг, т.е. однократная "укладка жестких участков вторичной структуры на суставах Pro", и динамический "фолдинг", т.е. по существу функционирование пико-механизмов. Я даже показал динамические модели белков, демонстрирующие этот динамический "фолдинг":



{kind=link}
Однако Виктория пишет не об этом "фолдинге третичной структуры", а именно о мистическом фолдинге вторичной структуры, когда этот мистический "фолдинг" должен сначала разрушить построенные рибосомой "шаблоны", например, левую спираль с углами между аминокислотными остатками в 120 градусов, разорвать все водородные связи n ... (n+3), изменить все углы со 120 на 87 градусов и замкнуть заново все водородные связи уже по схеме n ... (n+5). Вот о чём дискуссия, а не о том, что угол между альфа-спиральными участками может меняться на Pro.
2017.12.11 10:46:43
aest пишет:
Напоминаю слова Виктории:
"Кстати фолдинг это не кардинальное изменение закодированного структурного шаблона, а лишь его "косметическая" оптимизация."
Кушелев: А за этими словами кроется разрушение всех водородных связей, изменение всех углов и создание всех водородных связей заново. Вот такая "травматическая косметика"
. . . . . Было . . . . . . . . . . Стало
2017.12.11 10:58:08
aest пишет:
Если я не ошибаюсь, альфа спираль у Виктории получается в результате фолдинга.
Кушелев: Совершенно верно! Рибосома по композиционному коду строит сначала 310-спираль (по Виктории):
310-спираль альфа-спираль
после этого мистический фолдинг (будем называть вещи своими именами) разрушает все водородные связи n ... (n+3), меняет все углы между аминокислотными остатками с 120 градусов (3 АО на виток) до 100 градусов (3.6 АО на виток), после чего создает все водородные связи заново. "Травматическая косметика" в двух словах
2017.12.11 11:35:39
Victoriy пишет:
А как насчет Вашего обобщения частного случая лизоцима до всей когорты белков? Это ли не грубейшая философская ошибка, ведь дисульфидные мостики имеет менее 1 % белков.
Кушелев: Давайте разберёмся. Если 1% белков строится по композиционному генетическому коду, то какой вывод можно сделать о принципах сборки белков? Могут ли белки, в которых есть дисульфидные мостики строиться по иным принципам, чем остальные белки? Может ли альфа-спираль собираться по композиционному коду, а пи-спираль без композиционного кода?
Если уж композиционный код обнаружен, значит он работает для всех белков рибосомальной сборки. Не может же рибосома "тут работать, а там не работать". Это же абсурд!
Конечно есть нюансы. Рибосомы митохондрий работают по слегка другой таблице композиционного кода:

Более того, в некоторых организмах используется своя таблица генетического кода, но это нюансы. Если рибосома работает, то она работает одинаково при сборке любых рибосомальных белков.
2017.12.09 13:54:27
Victoriy пишет:
что Вы носитесь с этими дисульфидными мостиками лизоцима. Никто же не спорит о закодированности структуры белка в разной степени сохраняющейся в конформации белка после фолдинга последнего.
Кушелев: Если пространственная структура сохранилась в циклах лизоцимов на все 100%, то фолдингтне нужен.
2017.12.09 14:02:18
Victoriy пишет:
для лизоцима можно предположить замыкание находившихся по соседству в пространстве (а не в линейной последовательности аминокислот) SH-групп цистеинов в дисульфидные мостики ещё на этапе синтеза его структурного шаблона, что способствовало сохранению пространственного взаиморасположения КЛЮЧЕВЫХ узлов в этом ферменте (в частности активного центра).
Кушелев: Так по соседству атомы серы могут оказаться только в одном случае. Если все остатки установлены строго по композиционному коду. Случайно это невозможно. Для одного цикла из 22 АО вероятность случайного "расположения по соседству" равна 1/30 000 000 000, а для всех 6 циклов разных лизоцимов эту вероятность нужно ещё в 6-ю степень возвести. Получится (1/30 000 000 000)^6 = 1,37e-63 Просто нереальная вероятность.
2017.12.09 14:08:24
Victoriy пишет:
Гомологичные лизоцимы унаследовали найденный природой ход своего уникального сворачивания для формирования активного центра,
Кушелев: Если структура в окончательном варианте построена по коду, её уже невозможно свернуть. Она уже "свёрнута". Вы пытаетесь "фолдингом" положить в карман то, что там уже лежит.
В процессе эволюции композиционный код подстраивался под физико-химические взаимодействия, поэтому после денатурации белки иногда могут полностью ренатурировать. Но рибосома по композиционному коду собирает их не денатурированными, согласитесь. Известно же, что рибосома собирает белки быстро, а ренатурируют они медленно. Так что фолдинг тут неуместен.
2017.12.09 14:12:55
Victoriy пишет:
остальные несущественные детали
Кушелев: Кстати, о "несущественных деталях". В процессе исследования циклов разных лизоцимов, которые замыкаются через дисульфидные мостики, мне удалось обнаружить, что эти самые дисульфидные мостики возникают не только между цистеинами, но и между цистеином и метионином! Это научное открытие тоже прошло незамеченным, а зря...
2017.12.09 14:39:47
Victoriy пишет:
моя программа Молекулярный конструктор по коду AAG построит шаблон правой спирали полилизина, а по коду ААА - соответственно левой спирали. А далее виртуальный фолдинг с поиощью МД придаст им вид альфа- и пи-спиралей, если конечно их устойчивость выиграет в противостоянии с влиянием микроокружения. Вы же в курсе, что закодированные спирали сохраняются в конформации белка в лучшем случае на 80-90%, а в худшем - на 10-20 %.
Кушелев: Итак, в процессе дисскуссии выяснилось, что после исправления программы от Prosolver вторичные структуры полилизинов, построенных по кодам n(AAA) и n(AAG) по Соколик и Кушелеву полностью совпали:
Однако, Виктория Соколик утверждает, что рибосома соединяет водородными связями остатки левой спирали через 3 АО, а потом некий "фолдинг" разрушает все эти водородные связи и создаёт заново, но уже через 5 АО. Согласитесь, что это абсурд.
2017.12.09 14:52:15
Алгоритм определения вторичной структуры полилизина у Вас оказался правильный, т.е. точно такой же, как у меня. Просто в программе от Prosolver он не был реализован.
Осталось исправить таблицу углов, т.е. чтобы по коду пи-спирали строилась именно пи-спираль, а не полуфабрикат, который потом нужно переделывать в пи-спираль
2017.12.09 17:43:44
Victoriy пишет:
это не совпадение с Вашими моделями, а соответствие экспериментальным данным.
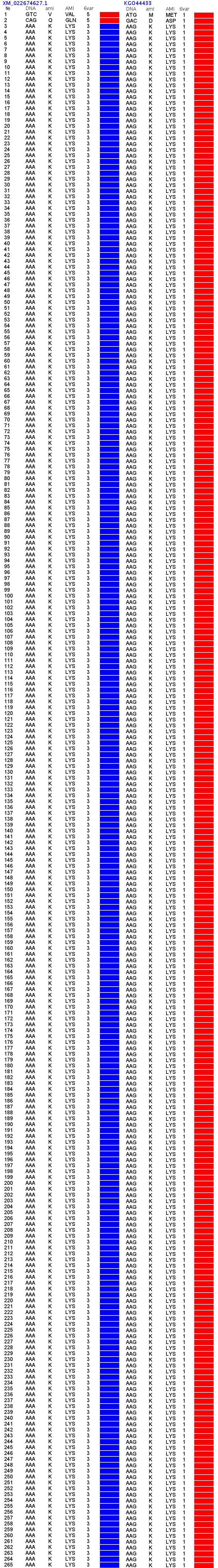
Кушелев: Я бы рад ошибиться и признать свою ошибку, особенно, если Вы сможете показать экспериментальные данные по белку: XM_022674627, который является прямой пи-спиралью длиной 263 аминокислотных остатка, т.е. 263/4.1=64 витка прямой пи-спирали.
И это при том, что официально считается: http://info-farm.ru/alphabet_index/p/pi-spiral.html
Цитата: Длинная ливозакручена π-спираль вряд наблюдается в белках
Конец цитаты.
https://ru.wikipedia.org/wiki/%D0%92%D1 … 1%80%D0%B0
Цитата: π-спираль, или спираль 516, — спираль с широкими витками, в результате в центре спирали остаётся пустое пространство. В белках встречается редко, обычно не более одного витка.
2017.12.11 02:03:52
aest пишет:
Виктория утверждает, что замыкание происходит во время синтеза структурного шаблона, а потом происходит фолдинг.
Кушелев: Вы точно ничего не напутали? Если дисульфидный мостик замкнулся, то пространственная структура белка зафиксирована. Это всё равно, что закрыть дверь на ключ.
Гипотеза фолдинга заключается в том, что из рибосомы выходит типа линейная цепочка аминокислотных остатков, которая сама, т.е. уже без рибосомы, сворачивается в объёмную конструкцию.
А я на примере циклов лизоцимов показал, что углы между аминокислотными остатками запрограммированы в таблице композиционного генетического кода, т.е. рибосома по композиционному коду сразу собирает вторичную структуру белка. Например, если код n(AAG), то из рибосомы выходит прямая альфа-спираль с водородными связями между n-ой и n+4-ой пептидными группами. Если код n(AAA), то выходит прямая пи-спираль с водородными связями между n-ой и n+5-ой пептидными группами. Сворачивать уже ничего не требуется. Либо нужно сначала разломать закодированную вторичную структуру, а потом снова сделать, что абсурдно, согласитесь. Открытие композиционного кода опровергло гипотезу фолдинга. Другое дело - укладывание вторичной структуры белка в третичную. Если между спиральными участками попадаются суставы Pro, то спиральные участки могут двигаться, как кости скелета на суставах. И тут уже вступают в действия силы притяжения и отталкивания между участками спиралей. Этот процесс вполне можно назвать фолдингом, но ... фолдингом уровня третичной структуры, которая складывается из жестких спиральных (и не очень) участков вторичной структуры. Циклы лизоцимов - это законченные третичные структуры, которые фолдингу не подлежат, т.к. дисульфидный мостик уже замкнулся и зафиксировал пространственную форму. И случилось это в процессе рибосомальной сборки, а не в процессе мистического фолдинга уровня вторичной структуры. Ведь дисульфидный мостик замкнулся уже на уровне геометрического алгоритма, т.е. композиционных кодов и композиционных углов. Это значит, что никаких взаимных движений на суставах Pro не было. Как построили, так и стоит.
Поймите, что нельзя построить (фолдингом или чем угодно) то, что уже построено. Либо надо ломать и строить заново, что абсурдно.
2017.12.09 13:35:57
Victoriy пишет:
и левая спираль или поворот (NNА)
Кушелев: А это уже ближе к делу. Значит Ваша программа по коду AAA действительно построит не правую, а левую спираль. Но реальная пи-спираль имеет 4.1 аминокислотных остатка на виток: http://info-farm.ru/alphabet_index/p/pi-spiral.html .
Аминогруппа одной аминокислоты формирует водородную связь с C = O группой аминокислоты на пять остатков ранее
А это значит, что Ваш "фолдинг" должен разорвать все водородные связи с третьими аминокислотными остатками и создать новые водородные связи с пятыми аминокислотными остатками, что сами понимаете, нереально.
По коду AAA рибосома сразу ставит аминокислоту так, чтобы водородная связь замкнулась на пятый аминокислотный остаток. И не надо ничего переделывать
Но самое интересное, что пи-спираль в Вашей программе так же однозначно закодирована, как и в моей программе Пикотех, т.е. в Вашей таблице AAA - тот же композиционный код пи-спирали, а код AAG - тот же композиционный код альфа-спирали.
Вы просто ввели в заблуждение отсутствием вторичной структуры в программе от Prosolver:

Пи-спираль по коду AAA (Кушелев) и отсутствие вторичной стуктуры по коду AAA (Соколик)
Я так понял, что эту ошибку Вы частично исправили в программе "Молекулярный конструктор", т.е. теперь по коду AAA программа показывает не отсутствие вторичной структуры, а левую спираль, но ещё не 4.1 аминокислотных остатка на виток, а 3. Но это тоже дело поправимое. Ведь в следующей версии можно изменить 3 на 4.1. И не нужно никаких чудо-"фолдингов" для превращения всех неправильных углов пи-спирали в правильные
2017.12.09 13:33:17
Victoriy пишет:
На основе структурного шаблона правой спирали в ходе фолдинга могут быть смоделированы все три основных разновидностей правых спиралей: 3/10-, альфа- и пи-.

Кушелев: Так-так-так... Пи-спираль вообще-то левая, 4.4 остатка на виток. Но я уже писал, что бог с ними, с этими переделками 310-спирали в разные спирали белков. Вы скажите, откуда "фолдинг" узнает, во что переделывать "310-шаблон". Ведь информация о том, что полилизин будет пи-спиралью, содержится в кодах: n(AAA), а информация о том, что полилизин будет альфа-спиралью, содержится в кодах n(AAG). В шаблоне информации типа AAA/AAG уже не содержится. Верно? Так откуда "фолдинг" узнает, во что переделывать шаблон? Где информацию брать?
Рассмотрите пи-спираль полилизина, построенная по триплетам AAA и альфа-спираль полилизина, построенная по триплетам AAG.
2017.11.24 11:26:02
фолдинг этих самых структурных шаблонов в энергетически устойчивую функциональную конформацию
Кушелев: Уважаемая Виктория! Как Вы прокомментируете открытие программных спиралей белковых молекул?
Здесь Вы видите 19 вторичных структур поливалина. На схеме вторичной структуры представлены фундаментальные спирали:
альфа-, 310-, пи-, бета-
и программные:
14-, 35-, 352-, 144-, 25-, 22225-, 411412-, 255-, 2444-, 1144-, 11444444-, 23-, 335(коллагеновая)-
Помните, как мы с Вами дискутировали по поводу лизиновых "хвостов", т.е. участков пи-спиралей длиной более 10 аминокислотных остатков? Вы ещё писали, что это всё гипотетические структуры...
А как Вы прокомментируете тему: "Рекорды сверхдлинных спиралей белковых молекул"?
Там встречаются фундаментальные спирали (альфа-, 310-, пи-, бета-) длиной более 3000 аминокислотных остатков. И программные спирали, в т.ч. фрактальные более 100 периодов, например, по 35 аминокислотных остатков в периоде, т.е. более 3500 аминокислотных остатков в спирали.
Если бы была верна гипотеза фолдинга, то зачем было бы кодировать все эти спиральные структуры?
Антигомология = программная аллотропия опровергает гипотезу фолдинга. Согласны?